

 UK ravimiamet ei kontrollinud üldse Pfizeri vaktsiiniuuringu andmeid, ilmselt ei teinud seda ka Eesti ravimiamet
UK ravimiamet ei kontrollinud üldse Pfizeri vaktsiiniuuringu andmeid, ilmselt ei teinud seda ka Eesti ravimiamet
 16. märts 2022 kell 16:37
16. märts 2022 kell 16:37

Foto: Canva
Kirjutasime möödunud nädalal Austraalia teabevabaduse seaduse kaudu ilmsiks tulnud skandaalist, milles selgub, et Austraalia terviseamet kiitis mRNA vaktsiinid heaks ilma regulatiivse kontrollita. Täpselt samad asjaolud tuvastati Ühendkuningriigi vastavaid dokumente analüüsides: kuigi eksperdid siin- ja sealpool lompi teatasid nagu ühest suust, et eksperimentaalsed mRNA vaktsiinid on ohutud, siis tegelikkuses ei kontrollinud selleks ette nähtud asutused ja ravimiregulaatorid nende preparaatide ohutust üldse.
On selgunud, et Ühendkuningriigi meditsiiniline reguleerija ei kontrollinud kunagi Pfizeri COVID-19 kliiniliste uuringute andmeid, rakendades sama praktikat nagu Austraalia, kus ravimiregulaator (Therapeutic Goods Administration, TGA) kiitis Pfizer-BioNtech mRNA eelravimi, uurimisravimi heaks, kuid hiljem selgus, et neil pole isegi olemas Pfizeri vaktsiinide heakskiitmise protsessiga seotud dokumente.
Pfizeri esitatud kliiniliste uuringute andmete valideerimine
Kuigi valitsused ja staararstid/eksperdid räägivad endiselt, et vaktsiinid on ohutud ja tõhusad, siis ülemaailmsed andmed, eriti Ühendkuningriigis ja Iisraelis (aga ka väiksemates riikides nagu Island ja Gibraltar, kus inimkatsetes osalemise määr läheneb 100%-le, kuid juhtumid on endiselt tõusuteel) on näidanud pettumust valmistavat reaalsust päris maailmas, erinevalt Pfizeri väidetest.
Arvestades Pfizeri seaduserikkumisi täis ajalugu (altkäemaksud riigiametnikele, ravimite vale klassifikatsioon, andmete fabritseerimine), oli hädavajalik, et ravimiregulaatorid kontrolliksid ravimitootjate esitatud andmeid enne heakskiitmist. Austraalia TGA seda ei teinud ning Ühendkuningriigi MHRA (Medicines and Healthcare products Regulatory Agency) samuti mitte.
Teabevabaduse seaduse alusel esitatud päring
2021. aasta juunis esitati MHRA-le teabevabaduse taotlus, milles otsiti dokumente Pfizeri (BNT162b2) ja AstraZeneca (ChAdOx1) COVID-19 geeniravimite ajutise loa kohta.
Teabenõude vastuses märgitakse, et „sponsoril, st Pfizeril, oli juurdepääs algandmetele“, ning MHRA suunas taotleja varasemale komitee inimravimite komisjoni koosolekule (CHM), mis toimus 24. detsembril. 2020. Selle koosoleku protokollist selgub, et CHM-ile oli öeldud:
„MHRA analüüsi vaheanalüüsi algandmete tulemused näitasid, et VE oli 91% (CI 63, 98) ajavahemikul 14. päevast pärast esimest annust kuni anti teise annuseni. Arutati ka Public Health Englandi sõltumatu analüüsi tulemusi Pfizeri täieliku andmekogumi kohta.“
(VE – vaktsiini efektiivsus; CI – usaldusintervall – toim.)
CHM koosoleku kokkuvõttev dokument on siin.
Kuid UK teabevahetuse seadus (FOI) vastuste põhjal saab kinnitust oletus, et MHRA ei ole üldse Pfizeri algandmeid analüüsinud.
MHRA vastusest saab lugeda:
„Selleks, et selgitada meie varasemaid väiteid, kinnitame teile järgmist: kliiniliste uuringute patsienditasandi andmed esitatakse MHRA-le koos kõigi uute ravimite müügiloa taotlustega. Sponsorile volitust ei antud, kuna seda polnud vaja.“
Seejärel suunas MHRA teabenõude esitaja valitsuse veebilehtede avalikele postitustele COVID-vaktsiini lubamise kohta.
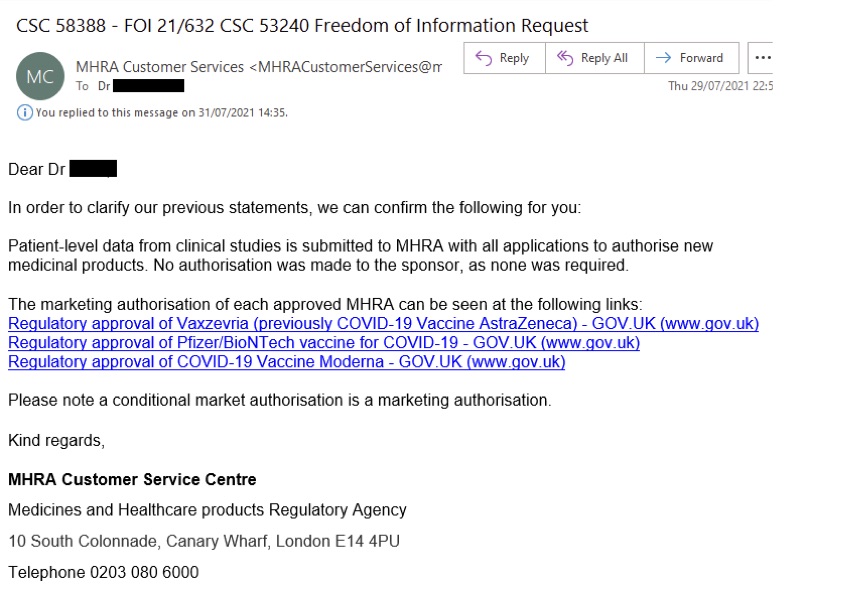
See vastus on selgelt vastuolus CHMi 24. detsembri protokolliga.
Arvestades, et MHRA oli teabenõude esitajale viidanud CHMi koosolekule kui tõendusmaterjali selle kohta, et “kliiniliste uuringute andmeid analüüsiti nii, nagu neid tehakse müügiloa saamiseks”, esitati 31. juulil 2021 veel üks teabevabaduse taotlus – seekord Public Health Englandile. Teine taotlus oli seotud PHE väidetava “sõltumatu analüüsiga … Pfizeri täieliku andmestiku kohta”, nagu tsiteeriti CHMi koosolekul.
Taotlus
Kirjutan, et taotleda kõiki dokumente, mis on seotud Pfizeri täieliku andmekogumi sõltumatu analüüsiga Public Health Englandi poolt, millele viidati 24. detsembril 2020 toimunud inimravimite komisjoni koosolekul.
Dokumendid peaksid sisaldama, kuid mitte ainult:
(1) analüüsi kokkuvõte
(2) analüüsi täielik kokkuvõte
Vastus oli kiire ja eitav.
PHE vastuses öeldakse:
„Vastavalt FOI seaduse jaotise 1 lõike 1 punktile a võib PHE kinnitada, et ta ei oma seda teavet.
Teie taotluses soovitatud analüüsi kokkuvõtet või täielikku kokkuvõtet ei ole.“
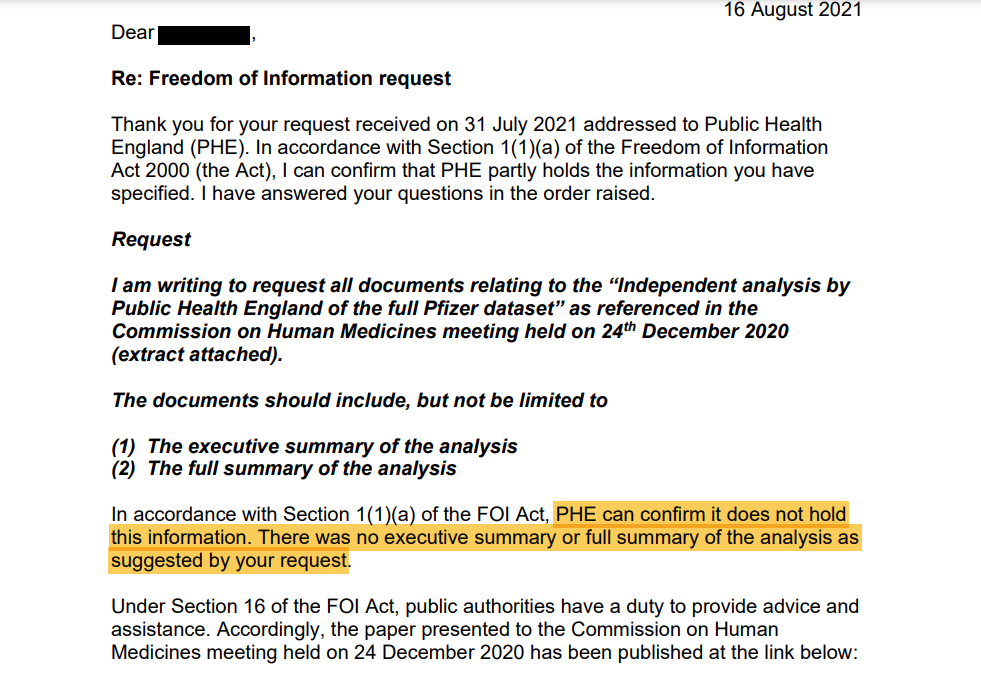
See tähendab, et PHE-l ei olnud juurdepääsu kogu Pfizeri andmekogumile.
Teisisõnu, MHRA (Ühendkuningriigi ravimiregulaator, mille ülesandeks on tagada uute ravimite ohutus) ei suutnud Pfizeri andmeid ise analüüsida. Lisaks väitsid nad, et andsid selle töö Public Health England (rahvatervise organisatsioonile), kes väitis kategooriliselt, et neil pole andmetele juurdepääsu, rääkimata andmete valideerimisest või analüüsist.
Inglismaa rahvatervise teabetaotlusega seotud dokument on saadaval siin.
Niisiis, see on neljast suurest ravimiregulaatorist (MHRA, TGA, EMA, FDA) teine, kes pole kunagi kontrollinud ega hinnanud Pfizeri imeandmete kehtivust.
Mis tekitab küsimused:
Vastused neile küsimustele on COVID-19 vaktsiinide tõttu rahvatervise ja avaliku turvalisuse seisukohast kriitilise tähtsusega.
Koroonavaktsiinide ohutusuuringute pettus on lääneriikide olematute heakskiitude seeria Archilleuse kand.
Mida väidab Eesti Sotsiaalministeerium koroonavaktsiinide turvalisuse ohutusuuringute kohta?
Avaldame Sotsiaalministeeriumi poolt meie lugejale saadetud teemakohalise e-kirja:
Tere!
Olete pöördunud Sotsiaalministeeriumi poole.
Vaktsineerimisega seotud informatsioon on kättesaadav veebilehel https://vaktsineeri.ee/
Vaktsineerimine on rangelt vabatahtlik.
Kõik Eestis kasutatavad vaktsiinid on Euroopas müügiloa saanud ja Euroopa Ravimiamet EMA on hinnanud need piisavalt ohutuks ja tõhusaks koroonaviirusega võitlemisel. Vaktsiine analüüsivad teadlased ja ametiasutused, kelle ülesanne on garanteerida, et need vastaksid kõikidele kehtivatele kvaliteedi-, ohutuse ja tõhususe nõuetele. Müügiloa saamise tingimusi pandeemia tõttu ei ole leevendatud ega ole tehtud järeleandmisi hindamiskriteeriumites (ohutus, kvaliteet, efektiivsus).
Silja Elunurm
upTIS teisese andmekasutuse valdkonna juht
Tervisesüsteemi arendamise osakond
xxxxxxxx | silja.elunurm@sm.ee
Sotsiaalministeerium
Suur-Ameerika 1 | 10122 Tallinn
6269 803| https://www.sm.ee

Esiteks: Sotsiaalministeerium eksitab ilmselgelt vastuses pöördumisele teadva kohustusega ravimite klassifikatsiooni osas, kui nimetab uurimisravimit, testravimit vaktsiiniks.
Teiseks: Sotsiaalministeerium väidab, et testravimid on ohutud ja tõhusad, selle väite on kummutanud teadus (tegemist on 3. faasi ravimiuuringutega), vaktsiinidest tingitud kõrvalmõjude ja surmade kohta on kogutud piisavalt tõendeid üle maailma.
Kolmandaks: Sotsiaalministeerium ei ole esitanud dokumente, et riigisisene pädev ametkond on kontrollinud DNA/mRNA testravimite, uurimisravimite ohtust ja tõhusust, mille alusel tingimuslik ravimiluba (EUA) riigisisesel tasandil ravimifirmadele (neli nimetust) on antud.
Neljandaks: Sotsiaalministeeriumi riigiametnik esitab faktiväite:
Euroopa Ravimiamet EMA on hinnanud need piisavalt ohutuks ja tõhusaks koroonaviirusega võitlemisel on ohtust kontrollinud.
Sotsiaalministeerim ei ole esitanud raportit, mis tõendaks, et Euroopa Ravimiamet [European Medicines Agency] EMA on viinud läbi kontrolluuringu ning kontrollinud selle käigus tegelikkuses COVID-19 DNA/mRNA testravimite (i) ohutust ja (ii) tõhusust enne EUA andmist.
Viiendaks: Sotsiaalministeerium väidab 11. märtsi 2022 vastuses päringule, et COVID-19 DNA/mRNA testravimitega tehtavas ravimikatses osalemine on vabatahtlik.
See väide ei pea paika!
Vastupidi: COVID-19 immuunsuspass (ka roheline pass) on kaudne sunnimeetod. Seda määratlust järgib ka meditsiiniõigus ja õiguskirjandus, see tähendab COVID-19 immuunsuspass on juriidiliselt määratletud kui kaudne sundimine.
Lõpetuseks:
1. Küsimus Eesti Ravimiametile:
Mille alusel DNA/mRNA eelravimid heaks kiideti ja kuidas suutis vastutav asutus, antud juhul Eesti Vabariigi Ravimiamet täita oma juriidilist kohustust testida uudse uuritava ravimi ohutust ja tõhusust?
2. Dokumentide nõudmine Ravimiametilt:
Palun esitage dokumendid, mis tõendavad COVID-19 DNA/mRNA testravimite ohutust ja tõhusust, mille alusel anti farmaatsiafirmadele Pfizer, Moderna, AstraZeneca ning Johnson & Johnson COVID-19 DNA/mRNA testravimite turustamisel erakorraline või ajutine kasutusluba riigisisesel ravimiturul.
Selle nõudega tegelevad Telegrami advokaadid, kes suhtlevad Ravimiametiga.
Toimetas Hando Tõnumaa
Allikad: lingid teksti sees
Foto: Canva


















Kommentaarid
Kommentaare lugeda ja kommenteerida saavad vaid Minu Telegrami tellinud kasutajad. Tellimuse esitamiseks kliki siia või logi sisse siit.